2.1. Overview¶
2.1.1. Goal and Perquisite¶
In this page is presented an overview of how  run. The aim is not to provide detailed information about how to use the code, this is given in to other tutorials, but to provide a general understanding of the software philosophy.
run. The aim is not to provide detailed information about how to use the code, this is given in to other tutorials, but to provide a general understanding of the software philosophy.
First, we present the general objective, then the different key objects that are used. Finally, what are the different step of a run and the general architecture of the directory and files.
2.1.2. Frog purposes¶
: FROm molecular dynamics to second harmoinic Generation
has been designed to calculate the second harmonic generation response of molecule in liquid phase from molecular dynamics and quantum calculation. It can be decomposed in two main functionalities.
First, is able to analyse trajectory obtained by Molecular Dynamics (MD). It aims to provide a user-friendly platform to perform complex structural analysis. For instance, you can easily compute the “molecular orientation” or “H-bond” of molecules.
Second, is designed to work with the QM software  in order to compute optical related properties within an electrostatic embedding. This should allow you to keep using your favourite MD software, to provide affordable dynamics, and still having various ways of approaching your optical observables.
in order to compute optical related properties within an electrostatic embedding. This should allow you to keep using your favourite MD software, to provide affordable dynamics, and still having various ways of approaching your optical observables.
The outputs are distributions: all the molecular quantities (molecular orientation, polarizabilities…) can be computed in function of the molecule position in the box, for instance with respect to the position in the MD box. This kind of discretization is called ‘geometrical selection’ thoughtout . The molecular property values in function of this geometrical selection is returned as “diagrams”. For instance, these diagrams can contain the hyperpolarizability distribution in function of the molecules position with respect to an interface.
2.1.3. Key Objects¶
Therefore, the bed-stone of is the package MDAnalysis, which is a powerful tool to read MD trajectory from various software and format. Once the trajectory is loaded, descriptions of every molecule have to be provided, so that can built electrostatic environment from the raw position of all the molecules for instance. Every molecule should be assigned to a Molecule Type (MT) which refers to a molecular library file in . For instance, the MT Water_TIP4P2005 or Chlore. The MT describes the function and property to use for the molecule assigned: what is the ‘mean position’ of a molecule, how to compute the ‘H-bond’ or how it generates electrostatic field in its neighbourhood.
Note
The name of the MT used to describe a given set of molecules can be different from the one given during the MD simulation. The assignment is done using the residue (molecule) number. See here for more details.
Hence, you may have to define your own MT: for a given analysis of an already available molecule, or because the molecule you used is not in the library. See here` for the available MT, and here to have more information about how to build your own MT. Please do not hesitate to contact us if you want to share your MT with us!

Once the molecules are assigned to one MT, you will be able to set what kind of analysis you want to perform for this MT.
Analysis are separated between the structural (like density, RDF…) and optical (polarizability, hyperpolarizability…).
The optical properties can be obtained using quantum calculation via the software where the other only requires analysis from the MD trajectory.
These properties are computed for every molecule within this MT (or not depending on the parameters) for every treated time step.
Results are stored for every molecule and time step.
At the end for the run, the results are regrouped into diagrams (which contain the results for every treated time step) and the diagrams are returned.
To summarise, for every MT define in your input, has a MoleculeType object associated. This object contains:
General information to describe these molecules and the parameters used for the different analysis: for the diagram and for the optical analysis.
A list containing all the molecules. Every molecule contains the result of the analysis required for every time step.
All the diagrams associated to every analysis. These diagrams will be averaged for every time step.
The other important object of is the Global Parameter (GP). It contains all the information impacting all the procedure, like the MD trajectory to read, the amount of time step… There is only one GP parameter for the whole run.
2.1.4. Steps¶
The software executes mainly 4 steps: the initialisation and the so-called first, second and third part – amphibian sometimes lack imagination for the names.
Warning
the following scheme describes the correct functionement, but not the good function names!
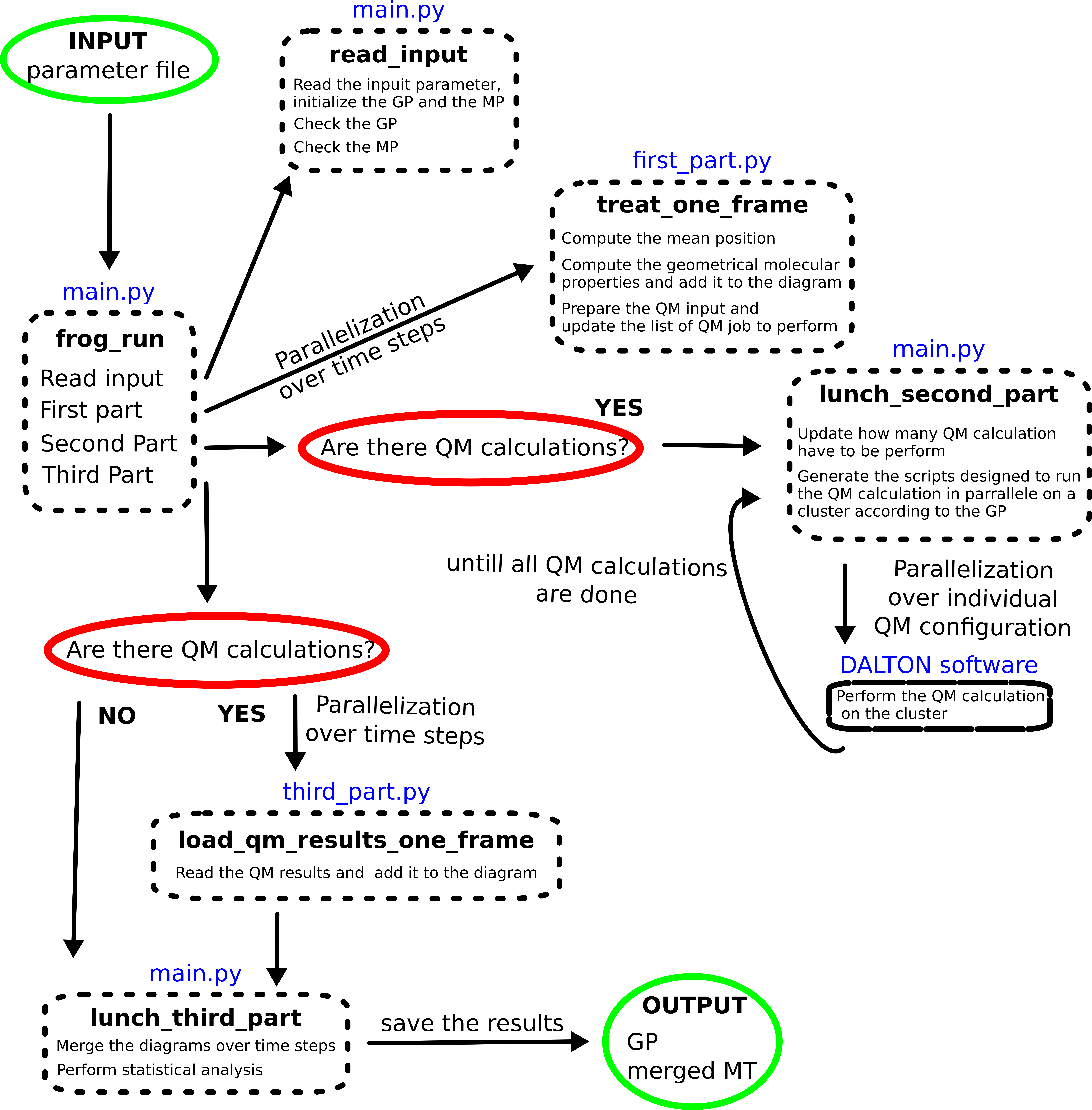
2.1.4.1. Initialisation¶
During this phase, the GP is initialised. checks many things to avoid unpleasant crashes later on. For instance, that the directories defined to store data are available (and that has the rights to write and read in these directories)… It will also print many messages to show what information it has understood and what will be performed.
It also initialises every MT object defined. Every MT object would also be checked. For instance, if the topology defined in the molecule module matches the MD trajectory.
Finally, the GP will be modified according to the analysis that should be performed for all the MT. It also check that all the MT objects have the requirement needed if some cross analysis have to be performed. For instance, that all the MT have a proper electrostatic description if a polarizable embedded QM calculation is required.
2.1.4.2. First part¶
All the structural properties (for instance the density or the molecular orientation) for all the time steps required are computed. Moreover, optical analysis where no QM calculation is needed are also computed. The diagram of these computed properties for all the time steps are also updated according to each molecule’s value.
Finally, if required, the input files are created. These files will be used in order to perform the QM calculations needed to extract optical properties. A unique list (for every time steps required) of all the QM run to perform is created.
The only output on the shell will be the time spend to treat each frame on the first core. It allows to estimate the time left before the end of the process.
When all the time steps have been treated, FROG goes to the second part. Note that this part can be run in parrallele.
2.1.4.3. Second part¶
If no QM calculations are needed, this part is skipped. Otherwise, in this part cluster-intended submission script are written in order to help you compte the QM configuration prepared in the first step. More information can be found here. To sumarize, here is the procedure:
Preparation of QM calculation submission
If it is the first time that the second part is called, then all the QM calculation prepared during the first part should be performed. For all the time steps,
reads the QM calculation that should be performed and creates a list merging all of them. This list is saved in a file named job_to_perform.p.Using this list of the QM calculation to perform and accordingly to parameters provided by the user,
prepares submission files to perform these QM calculations. These files are “cluster-intended” and aim to perform a lot of (independent) QM calculations at the same time. prints on the shell what files it has prepared at the end of this second part, and then stops.Execution of QM calculation
only prepares the submission file: the user has to launch them manually. Before launching them, we warmly recommend to have a look to the submission file prepared and to try several QM calculations alone to have an idea of the time needed. These manual verifications are there to avoid cataclysmic request to the cluster and to estimate the time needed for these calculations. If you are unfamiliar to cluster good behaviour, do not hesitate to ask for advice before submitting thousands of calculations. Here (TODO) are some remarks and advice regarding hyperpolarizability prediction in liquid-like phase, especially regarding the simulation time needed.Again and again
Depending on the parameters, all the QM calculation can be sent or not. Moreover, you might have some crashes during some QM runs. That is why this second part is designed to be recalled so that you can resubmit the calculation left to perform.
To do that, you can skip the first part while conserving the previous results. In this case, the second part of the run is not called for the first time, and
only updates the list of QM calculation to left to do. Then, if there is QM calculations to perform, it prepares the new submission file and then stops. The user has to perform these QM calculations, and launch again .Condition to go to next part
When the second part is called while all the QM calculations have been performed,
does not prepare any submission file and goes to the third part.
2.1.4.4. Third part¶
If any optical analysis needing QM calculation have been performed, reads the results of the QM output. The observables are stored for every molecule concerned by a QM calculation for all the time steps. Then, as during the first part of the run, the optical diagrams are updated according to each molecule’s values.
Finally, when all the time steps are treated, regroups all the time step results into one file – L_moleculetype_result.p by default. This object is very similar to the list of the MTs defined at each time step: it is the list of all the MT at the initial time step with the merged diagrams. Therefore, it contains the molecule values for the initial time step so you can have a look. However, the diagram available in this object contains the information of all the time steps. More information regarding the data analysis is available HERE.
2.1.5. Frog Files and Directory Structure¶
To conclude this overview, here is a presentation of the files used by :
- The MD files:
The topology and the trajectory files from your MD run. In this wiki we do not present at all how to perform a (meaningfull) MD simulation. See this part for the supported MD ouput.
- The parameter file:
All the definitions for the run are set in this specific file. The construction of this file is described in the following tutorials.
- The molecular module files:
The properties of a given MT are defined in such module. They can be viewed as “toolboxes”, i.e. a file that contains the function used to calculate the properties a specific kind of molecule. A library of molecular module is already available for basic molecules. However, we are aware that you may need to modify an existing molecular type or write a new one: this page presents how to do that.
- The template for the QM submission file:
If any QM calculation should be performed, a template used to submit a QM calculation should be given. See this page for more informations.
During the run, several files will be generated into several directory. :ref:` Some GP attributes<gp_directory_management_page>` control where are located these files:
To save the MT for every time step
By default called Molecule_times. At each time step, a file containing the list regrouping all the MTs (named L_moleculetype in the software) is stored in a time-labelled file L_moleculetype_t.p, where “t” is the time step.
The trajectory corresponding to every time step is also saved there if required.
- To save the QM input and output file
By default QM_Simulations. For every MT which requires QM calculation, create a directory where all the input files and the output of
are stored.- To save the submission files and their output
By default Submission_script. If QM calculations have to be performed, the scripts used to launch the jobs to the cluster are stored in this directory.
At the end of the run, the results are given in two files:
One recording the Global Parameter used, saved as GP.p.
One with the diagrams regrouping properties at all the time step values, saved as L_moleculetype_result.p by default.
Note
In order to help the user to use