4.1. How to create MT¶
The Molecule Type objects are 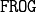 backbone. In python, this is an object of the class MoleculeType. There are as many MT object as different type of Molecule Type.
backbone. In python, this is an object of the class MoleculeType. There are as many MT object as different type of Molecule Type.
Note
In principle you may define several times the same type of MT (for instance Water_TIP4P) as long as every molecule of the MD trajectory are assigned to exactly one MT. However this has never been tested extensively.
The routine described as follow should be repeted for every MT declared. At the end, all the MT sould be stored in a list named L_moleculetype. The order of the MT in this list should not matter.
4.1.1. Initialization¶
To initialize any new MT for your run, you should use in the parameter file:
myMT = frog_class.MoleculeType(GP, molecule_type_name, where_are_molecules)
The GP is the usual General Parameter object defined first in the parameter file.
molecule_type_name is the name of this MT. The name of the MT should refers to an existing molecular library file. This file will be used in order to perform some molecular specific operation, like computing its “mean position” or return an “electrostic description”.
Finally, where_are_molecules MT is the ways to link a molecule of the trajectory to this molecule type:
where_are_molecules = ’all’ : all the molecule of the topology file are assigned to this MT.
where_are_molecules = [n,m] : the molecule number, with respect to the topology labeling, from n to m included are assigned to this MT. For example where_are_molecules = [1, 1700] include molecule numbered 1 to 1700 to the MT.
Note
Today no discrete way to assign MT to molecule (like [1, 4, 18]) is provided.
When reading this MT definition, runs a safeguard: it checks if the MT description required fits with the atoms present in each “molecule” of the topology file. Indeed, a molecular library file expects a specific atom order. For instance, the molecular type ’Water_TIP4P’ – defined in the molecular library file Water_TIP4P.py, expects the atomic order to be: Oxygen, Hydrogen, Hydrogen.
For MD countability sake, does not take into account the atom name defined during the MD. Thus, during this check, it makes sure the atom number matches for all the molecule defined to be part of this MT. Moreover, it checks that no molecule are “scattered” by verifying the atom distance between each other according to the molecular library file requirement – using if available the PBC conditions. prints the expected order of atom while running this check. If you want to change this definition, you have to change the molecular library file. See Here for more details.
4.1.2. Define Diagram¶
After this definition, the analysis you want to performed have to be declared using diagrams. For more information, see this page, here is a typical example:
input_for_diagram_1 = ['density', 'Plane_xy', 100]
input_for_diagram_2 = ['molecular_orientation', 'Plane_xz', [50, 100], 'independent']
L_diagram_analysis_to_perform=[input_for_diagram_1, input_for_diagram_2]
moleculetype.read_diagram_input(GP, L_diagram_analysis_to_perform)
4.1.3. Optical Analysis¶
Eventhough you have not decalred any ‘optical’ analysis, the optical parameter should be defined. In the minimal case, use:
moleculetype.read_optic_properties_input(GP)
For more information about how to declare the optical parameter, see this page.
Todo
a faire une fois la partie optical analysis d avance.
4.1.4. End the definition¶
Once the parameter for the diagrams and the optical analysis passed, you have to end the definition of the MT by the line:
myMT.end_initialize(GP)
L_moleculetype.append(myMT)
Many check are performed to make sure the parameter passed to the MT make sens. Crashes may happen later on, but this step try to avoid many of them. If you have required incompatible set of parameter, you should be given some information to help you fix that at this step.
Note
A final check before the real begining of the run is made once all the MT declared. The idea is to make sure that the code will not crash after hours of calculation if obvious mistake can be dectected early on.
The list of the MT, L_moleculetype, will be created for every time step and stored in the GP.dir_mol_times directory as pickle file. If you want to open any of these list at any time step, use the “pickle” module in an python environment. For instance, for the time step 3, open the L_molecule_type_3.p file. See the HERE more informations.