1.2. Numerical notes and requirements¶
1.2.1. OS compatibility¶
 has been designed to run on an UNIX environment. Tests on Debian and Ubuntu have been performed. No particular reason would lead to crashes for other UNIX version. However, please contact us if it is so.
has been designed to run on an UNIX environment. Tests on Debian and Ubuntu have been performed. No particular reason would lead to crashes for other UNIX version. However, please contact us if it is so.
Preliminary tests were performed on iOS: no problems were detected once the Python environment was created with the correct packages.
Tests has been made on Windows using the Anaconda environment. In such cases you may have trouble performing the regular tests.
1.2.2. Python packages¶
is written in Python3. It uses very standard libraries: os, sys, shutil, copy, datetime, time, pickle, importlib, multiprocessing, functools, itertools and numpy.
It also needs MDAnalysis and pytim.
1.2.3. Compatible MD software¶
works with the same topology during the whole simulation. No test has been made for MD simulation with varying number of particles nor chemical reactions. Therefore, the time step corresponding to the topology file does not matter as long as it is relevant with the trajectory encoding.
In order to use the MD trajectory files, uses the python module MDAnalysis. Therefore, FROG should be able to work with as many MD softwares as MDAnalysis can handle. However, here are the known successful attends until now: LAMMPS and NAMD. Below are listed how you should write the topology/trajectory line in these MD softwares. Please do not hesitate to let us know yours!
1.2.3.1. LAMMPS¶
Trajectory and topology files are a combination of .dcd and .data formats. In the FROG input file, use:
GP.MD_file_type = ’LAMMPS’
In LAMMPS, use:
dump dumpdcd all dcd your_dump_frequency mytrajectory_file.dcd
write_data mytopology_file.data
to write the trajectory during your MD run and the topology at the end of the simulation respectively. See also https://lammps.sandia.gov/doc/dump.html and https://lammps.sandia.gov/doc/write_data.html
1.2.3.2. NAMD¶
Trajectory and topology files are a combination of .dcd and .psf formats. In the FROG input file, use:
GP.MD_file_type = ’LAMMPS’
In NAMD, use:
DCDfile trajectoryfilename
DCDfreq X
to write the trajectory during your MD run. See also https://www.ks.uiuc.edu/Research/namd/2.9/ug/node12.html .
1.2.4. Compatible QM software¶
Only  V.2018 and V.2020 has been tried until now. No other QM software would be tested soon as the whole electrostatic emmebbeding procedure has been built around the PE formalisme of .
V.2018 and V.2020 has been tried until now. No other QM software would be tested soon as the whole electrostatic emmebbeding procedure has been built around the PE formalisme of .
1.2.5. Parallelisation of FROG execution¶
To speed up the execution of , the parallelization is made over the time steps to treat. Each cores will have equal number of time steps to treat during the first and the third part of the run. In the input file, use:
GP.nbr_parra = N
The python function “Pool” of the package “multiprocessing” is used to parralelize over “N” cores.
Of course, the easiest way for you to know how many cores you should use is to test by yourself. However, here are some advice that can help you guess:
The more cores you use, the less time step per cores should be performed during the first and the third part. Therefore, if your calculation requires a lot of ‘structural’ analysis, the more core, the less total time.
The third part run in parrallele for every time step to load the QM result. During the merging part,
Be aware that
Using GP.MD_cut_trajectory = True helps to reduce the RAM needed. You should use this option if you run
A botleneck can appear if many writting/reading operation is required (for instance when writing the dalton inputs in the first part) when runnning over many cores.
Here are some examples of parralelization analysis. The run consist on structural analysis only. The first part is the most time-consuming part. In the following figure is the total runing time in function of the number of cores used:
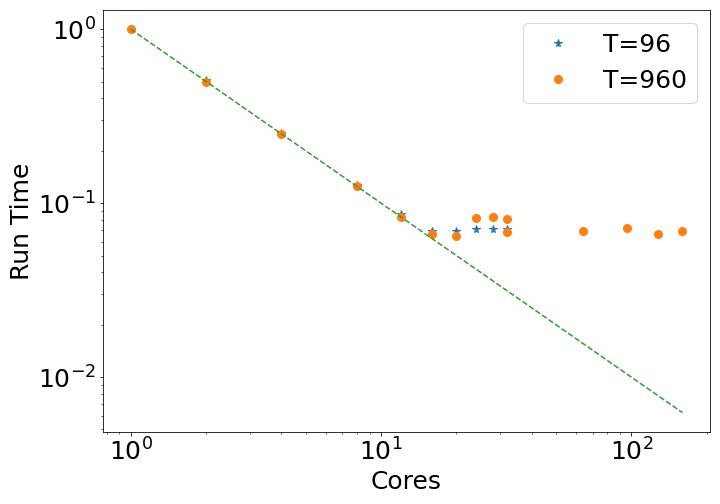
Note that each serveur contains 32 cores. Until 32 cores, only one serveur have been required where all the RAM has been reserved for . Two total number of time step has been tested: 92 or 920. The difference in normalized total run time (with respect to the value obtain with 1 cores) is negligeable for these 2 time step.
In this case, the total running time is inversly proportional to the number of core until 12 cores (the dashed line). However, after this point, the time needed for one core to treat one frame increases. The total running time still decreases until a plateau. Two problems can lead to such a result: not enough RAM available, or an overload writting-reading procedure.
Note
The point at 64, 96, 128 and 160 cores have been made with 2, 3, 4 and 5 serveur of 32 cores each. No clear amelioration is visible. Therefore, today you may consider that cannot benefit from running in parralelle over multiple serveur. Please note that it is not the case for the QM part, from which has been specifically designed to deal with QM calculation over multiple cores and serveurs.
1.2.6. RAM, Disk space and writing-reading procedure¶
1.2.6.1. RAM¶
can be quickly RAM-consuming. Indeed, the RAM most demanding part is the molecular information for all the molecules at a given time, and maybe the diagram if huge numbers of discretization points are asked. If parralelization over an important number of core is required, the total RAM used is:
< size of one frame > × < nbr of cores used by FROG >
Therefore, you may benefite from using a computer/serveur with large RAM.
Another important point is the trajectory file. If you are dealing with a trajectory with a lot of time step, you should use the option GP.MD_cut_trajectory = True . It will create a trajectory for every time step. This reduces dramatically the RAM needed to open the trajectory. Moreover, it prevent you from multiple access probleme if you are running in parralelle (several cores would use the same trajectry file if GP.MD_cut_trajectory = False).
1.2.6.2. Disk space:¶
There are two main occupancies of the disk by FROG : the molecular informations (orientation, polarizability…) for every time step that are contained in MT files and the QM input and output files.
The MT files are generated for each time step required and during the merging procedure deleted them?. Their size depends on the number of molecules and on the discretization asked for diagrams. The number of input files for the QM simulation can be very large quickly. All of these files are light (few hundred of bytes) apart from the environmental one (.pot and .xyz if asked) which can become large. We recommend to pay attention to the localization of these file: you may want to keep them to avoid redoing the QM simulations but they may take a lot of space. Today, no cleaning procedure regarding the QM input files has been implemented on FROG. You can find in Tutorials/Usefull_files/Bash_script a script named to_clean_directory_frog.sh which can be call using:
$ bash to_clean_directory_frog.sh
and deletes all usual output in the directory where this script is run.
1.2.6.3. Writing-reading procedure:¶
Be aware that if many servers are used at the same time to perform the QM run, the writing/reading process of files can very quickly become more time-consuming then the calculation itself. Therefore, we strongly recommend to write the QM input files in a disk well connected to the CPU – data-transfer speaking.
In the FROG input file, use to define the path to where the QM input/output should be placed:
GP.dir_torun_QM
Moreover, you can initialize the temporary files where DALTON will put some (large) files used for the QM run. In the FROG input file, use to define the path to these temporary files:
GP.scratch_dir
We recommend to use a /tmp or /scratch or any other disk space with a strong/robust data writing/reading procedure with the CPU. If you can use the RAM of the server it is even better. An example is shown in the tutorial.